The Aneuploidy Conundrum
Why Are Many Human Cancers Aneuploid when Aneuploidy Has Detrimental Effects on Human Development and Has Also Been Shown to Reduce Cellular Fitness?
1. Introduction
Aneuploidy refers to ‘the alteration of chromosome number that is not a multiple of the haploid complement’ [1]. Approximately 90% of solid tumours and 75% of haematological cancers exhibit some degree of aneuploidy [2]. Cancer is characterised by its capacity for over-proliferation. Aneuploidy, meanwhile, has long been associated with a decreased rate of cell proliferation in untransformed cells [3] and yet also predisposes to cancer [4]. The paradoxical nature of these findings has meant that the role of aneuploidy in cancer remains hotly debated, despite having first been investigated over 100 years ago by Theodor Boveri [5]. Studies on model systems are providing insight into this conundrum. By connecting the effects of aneuploidy on cellular fitness to those observed in cancer and human development at a multicellular level, one can begin to unravel this age old mystery [6].
2. Origins of Aneuploidy
No review of the aneuploidy conundrum would be complete without brief reference to how aneuploidy arises in cells. Aneuploidy is categorized into two forms. Constitutional aneuploidy arises from errors of chromosome segregation in meiosis of germ cell formation, and is present throughout the organism. Somatic aneuploidy is the result of such errors in mitosis, whereby only some cells in the organism are affected [7]. Spindle Assembly Checkpoint (SAC) mutations, aberrant cohesion or kinetochore interactions, and supernumerary centrosomes increase the rate of this chromosome mis-segregation [6]. The shared result is the production of lagging chromosomes during anaphase. Improperly separated from the ‘chromosome mass’, lagging chromosomes form their own micronuclei, or get damaged as a result of being trapped in the cleavage furrow [7].
3. Detrimental Effects of Aneuploidy on Human Development
Whilst the origins of aneuploidy are well characterised, the basis of its consequences are not. Long established in the literature is the causative association of aneuploidy with human death and disease; aneuploidy is the leading cause of mental retardation and spontaneous miscarriage [8]. Such is its detrimental impact on human development that ‘all monosomies and 20 out of 23 possible autosomal trisomies result in embryonic lethality’ [9]. Of the three constitutional trisomies that are viable in humans, only 5-10% of infants born with trisomy 18 (Edwards syndrome) or trisomy 13 (Patau syndrome) will live to 1 years old [10]. Developmental abnormalities shared between Edwards and Patau syndromes include cardiac defects, cleft palate and microcephaly in humans. [9]. Increased rates of congenital heart disease are also a phenotype of the third constitutional autosomal trisomy in humans; trisomy 21, (Down syndrome, DS) [6]. Whilst DS individuals may survive to adulthood, (perhaps attributable to the gene poor, small nature of chromosome 21), it is not without notable developmental defects. Marked hypotonia at birth, seizures [11], mental retardation, decreased fertility, and stunted growth are characteristic of DS in humans [6].
4. Aneuploidy Predisposes to Cancer
Central to this conundrum, predispositions to cancer associated with aneuploidy are detrimental to normal human development. Hasle indicates that DS individuals have a ‘600x increased risk for acute myeloid leukaemia (AML)’ and a ’20x increased risk for B cell precursor lymphoblastic leukaemia (ALL) compared to control children’. Yet DS individuals show a 50% lower probability of developing solid tumours than controls [15]. Mouse models suggest the additional copies of the Ets2 gene and Down Syndrome Critical Region1, DSCR1, on human chromosome 21, encoding a tumour-suppressor and a negative regulator of angiogenic signaling, respectively, might confer this gene specific, reduced risk in humans [16][17].
5. Sex Chromosome Aneuploidy: Escaping the Conundrum?
Defective (albeit less severe) human development and predisposition to cancer are observed also in the better-tolerated sex chromosome aneuploidies. As with DS, Turner syndrome females (45 XO), exhibit decreased adult stature and infertility [4]. A UK cohort study of 3425 women revealed that Turner individuals may be at increased risk for CNS, bladder and urethral cancers [18]. The same study also found the risk of breast cancer to be reduced. A corresponding phenotypic pattern is seen in Klinefelter syndrome (47, XXY). Men not only exhibit infertility and tall stature, a cohort study of 3518 men revealed a higher mortality from Non-Hodgkin lymphoma, breast and lung cancer [19]. In reviewing the effects of aneuploidy on human development, one might conclude that there is phenotypic overlap between viable human aneuploidies, be it cardiac, skeletal, neurological, fertility or cancer related. This implies that aneuploidy may have a general effect on human development, albeit with varying severity. Williams and Amon came to the same conclusion, hypothesising that this observation may be a reflection of the ‘dosage-sensitive genes required for neuronal, heart and skeletal developments’ carried by most chromosomes. They suggest also that this organism level phenotype may reflect a phenotypic effect of aneuploidy at the cellular level [1]. But is this also true of cancer cells?
6. Detrimental Effects of Aneuploidy on Cellular Fitness
Cellular level research on model organisms has made some progress in unraveling the basis of the effects of aneuploidy on human development [6]. Its findings on cellular fitness lie at the heart of this conundrum. The negative effect of aneuploidy on cell proliferation has been well documented in various model organisms. Systematic analysis of yeast S.cerevisiae disomes (carrying one or two additional chromosomes) by Torres and colleagues highlighted reduced cellular proliferation as a general effect in yeast [20]. Similar slowed growth phenotypes have been observed in mouse embryonic fibroblasts (MEFs) trisomic for chromosome 1, 13, 14 or 19 [21], and fibroblasts taken from DS individuals compared to controls [3]. The reduction in cell proliferation is thought to be partly attributable to the detrimental effect aneuploidy has on cellular protein quality control machinery [29]. The Gene Dosage Theory of aneuploidy states that ‘gains or losses of whole chromosomes immediately alter the dosage of hundreds of genes in a cell, thereby leading to an imbalance in critical proteins’ [9]. Transcriptome and proteome analysis in human aneuploid cells provides evidence for this; most protein products were expressed according to gene copy number levels [22]. Unlike for the sex chromosomes, no dosage compensation mechanisms existto correct gene copy number deviations for autosomes, and this may contribute to the more severe whole organism phenotype seen in autosomal aneuploidies [23]. Nonetheless, a subset of genes, enriched for gene-encoding proteins which form part of multi-protein complexes, did not show corresponding elevated levels in their protein concentration, resulting in stoichiometric protein imbalances [22] (Fig 1). It is thought that cellular protein quality control machinery, namely chaperones and protein destruction machinery (proteasome and autophagy systems) must ‘soak up’ the additional unpaired protein subunits encoded by the aneuploid chromosome [14]. Above a certain threshold, the protein homeostasis machinery is exhausted, and subsequent proteotoxicity, (the buildup of mis-folded proteins), places strain on the cell. ‘As little as a 0.1% increase in mis-folded protein places a burden on a cell’s protein quality control system and leads to a reduced cellular fitness’ in yeast [7][14][24]. Direct evidence of proteotoxicity in aneuploid budding yeast has been shown by Oromendia and colleagues. Hsp104:GFP fusion experiments indicated an increased likelihood of protein aggregate formation in aneuploid cells, in comparison to euploid controls [25].
Figure 1. Mechanism by which Aneuploidy Leads to Proteotoxicity.
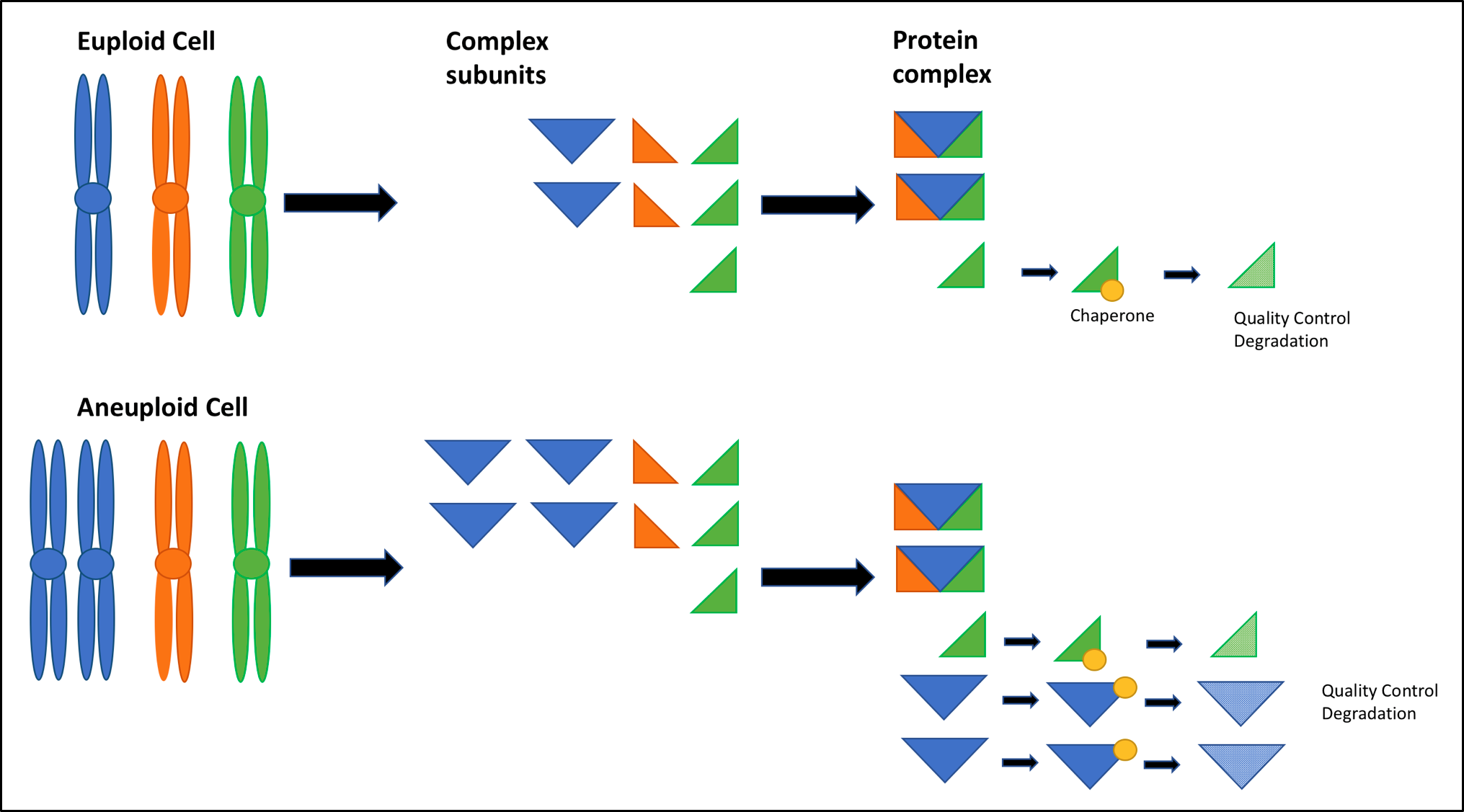
Excess protein subunits encoded by the aneuploid chromosome lead to stoichiometric imbalances. Unpaired subunits must be bound by chaperones to remain in solution, hindering normal chaperone activity until the subunits are degraded by the 26S proteasome or by autophagy; this places the protein quality control machinery of a cell under stress [14]. Proteins are thought to be altered by aneuploidy at the level of the interactome, too [26]. A study of patient tumour samples indicated that MET proto-oncogene amplification conferred resistance to EGFR inhibitors, erlotinib and gefitinib. At very high expression levels, MET is thought to activate kinases downstream of EGFR via many low affinity interactions partners, bypassing the anti-tumorigenic effect of EGFR inhibitors upstream [9][27]. Aneuploidy is thought to induce a similar over expression of proteins encoded by the aneuploid chromosome. Thus promiscuous protein interactions might be a mechanism by which aneuploidy alters the interactome to bring about the detrimental effect on cellular fitness in humans [26]. This mechanism may offer direct insight into the conundrum with respect to autonomous growth signaling seen in cancer. Alterations to cellular metabolism may act to reduce cell proliferation rates and cellular fitness [9]. The rate of biomass production was found to be decreased in aneuploid yeast strains, when compared to that of euploid controls [20]. Trisomic MEFs (produced via cdc20AAA mutations) exhibited increased ROS and lactate production, [28], as well as evidence for increased glutamine uptake and ammonium production in some MEF lines [21]. The same detrimental effects of aneuploidy on metabolism may also be seen in human aneuploidy. Metabolic alterations, particularly in the form of increased lactate production, is well established as part of the Warburg metabolism of cancer [29]. Perhaps aneuploidy in cancer cells contributes to this metabolic alteration [6]. Genetic instability associated with aneuploidy may further impact cellular fitness. Investigations of disomic budding yeast strains showed increased sensitivity to genotoxics, increased levels of DNA damage and chromosome mis-segregation [30]. Studies in mammalian aneuploid cells demonstrate that DNA damage and chromothripsis, associated with lagging chromosomes and micronuclei, respectively, can lead to extensive genetic instability and cell apoptosis. Thus, genomic instability likely contributes to the reduced cellular fitness associated with human aneuploidy [6]. Perhaps this genomic instability, thought to be key to cancer, represents a role of aneuploidy in cancer.
7. The Role of Aneuploidy in Cancer
Having detailed the current understanding of the detrimental effects of aneuploidy on human development and cellular fitness, one can begin to look to solving the conundrum with respect to cancer; does the aneuploid state confer benefits, causal or consequential, or does it prove detrimental to cancer cells? If aneuploidy were to confer benefits for cancer, perhaps this could offset the detrimental effect on cellular fitness, and go some way to resolving the conundrum. Evidence for a beneficial, causative role of aneuploidy stems from the observed predispositions to cancer associated in Down, Turner and Klinefelter syndromes [4]. Whilst correlation alone is very weak evidence for causation, the broad spread of viable aneuploidies that predispose to cancer surely does pose an intriguing biological hypothesis. Additional evidence for a beneficial role of aneuploidy in tumorigenesis is born of the idea that aneuploidy acts as a means of amplifying mutated oncogenes (Fig 2). Trisomy 8 is seen in 25% of chronic myeloid leukemia (CML) and 10-15% of acute myeloid leukemia (AML) [7]. The additional copy of MYC, carried on human chromosome 8, is thought to explain why this form of aneuploidy appears to be positively selected for in hematological cancers [7]. A similar relationship between aneuploid chromosome 3, the TERC proto-oncogene and cervical cancer is thought to offer further evidence for a beneficial role of aneuploidy in cancer [9].
Figure 2. Aneuploidy may be both beneficial and detrimental to tumorigenesis depending on the tumour cell context [30].
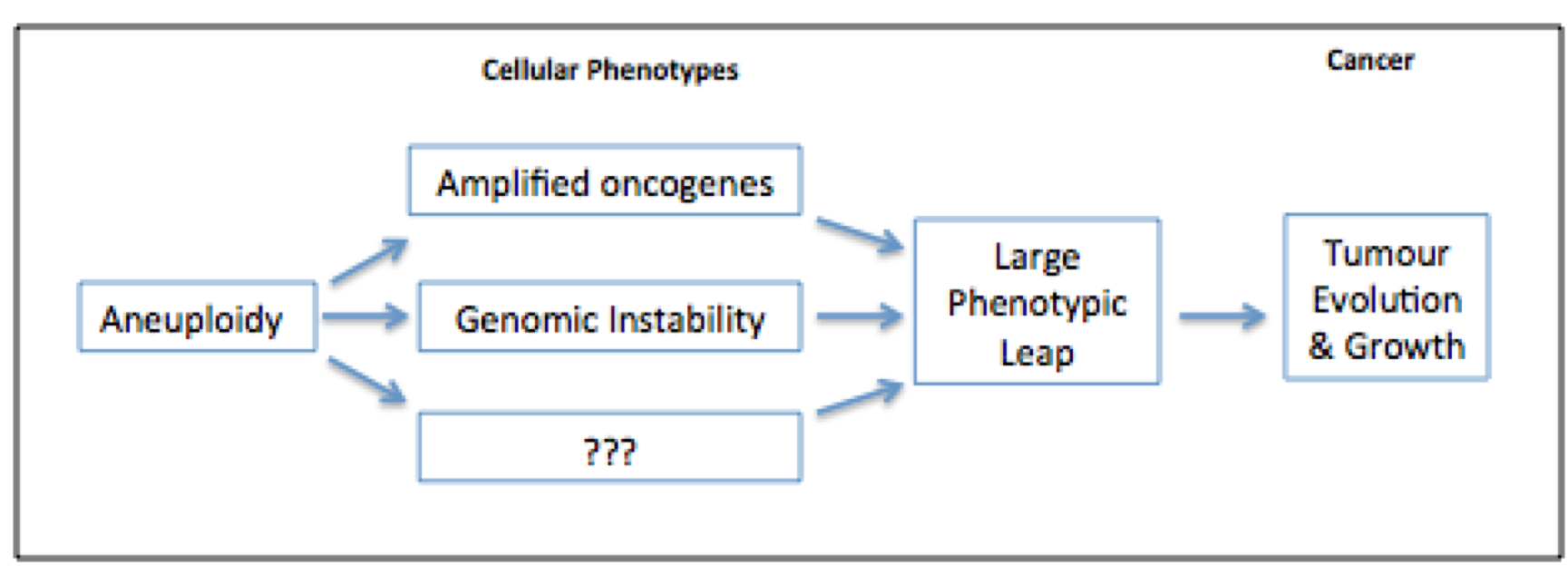
Increased genomic instability, as associated with aneuploidy, is thought be beneficial to tumour evolution; its associated karyotypic variation can hasten acquisition of growth-promoting mutations. Aneuploidy may be a particularly effective means of achieving this, as multiple mutations need to be acquired rapidly to allow adaption to new, stressful environments [9] (Fig 2). Evidence for this role of aneuploidy, specifically in the relapse and recurrence of tumours, has been shown in repressed KRAS-dependent tumours in mice. Over expression of MAD2 (known to induce chromosome instability and hence aneuploidy) and KRAS led to the formation of lung adenocarcinomas of a more aggressive nature than those induced by over expression of KRAS alone [32]. Sotillo and colleagues found that when KRAS expression was withdrawn from both types of tumours, reoccurrence occurred at significantly elevated rates in the MAD2 KRAS-dependent lung adenocarcinomas. Karyotypic analysis of the relapsed tumours showed them to be highly aneuploid with activation of multiple pro-proliferative pathways. This result is consistent with the idea that aneuploidy provides genetic diversity, which tumours are able to exploit in their evolution [24][32]. Amplification of proto-oncogenes and genomic instability are two possible cellular phenotypes of aneuploidy that may confer growth advantages to tumours. Others may well exist. Contrary to the above view, there is evidence that aneuploidy may be an unintentional consequence of tumorigenesis. Despite multiple structural and numerical karyotypic alterations being present in cancer cells, there are relatively few examples of specific forms of aneuploidy shared among a specific tumour type [33]. In addition, mutations of genes known to regulate chromosome segregation are rare in cancer [7], and even fewer of those aberrations have been shown to contribute to tumour formation [33]. What is more, genomic instability arises in cancer cells by means other than aneuploidy [24]. The loss of tumour suppressor Rb, for instance, has been shown to cause genomic instability by contributing to the accumulation of DNA damage, and defective chromosome condensation [34]. Similar findings have reported that the loss of APC also causes genomic instability by decreasing chromosome segregation fidelity [7]. These observations, coupled with the finding that aneuploidy alone is insufficient to cause tumour growth in culture, implies that aneuploidy may be neither necessary nor sufficient for the process of tumorigenesis [24].Overall, one can see the relationship of aneuploidy and cancer is rather complex. But the above conclusions that aneuploidy promotes or inhibits tumorigenesis need not be mutually exclusive. Classical theory of evolutionary adaption, first described by Kauffman and Levin in 1987, offers part of a solution to this conundrum [26][38] (Fig 3).
Figure 3. A Model For How Aneuploidy Promotes Tumorigenesis.
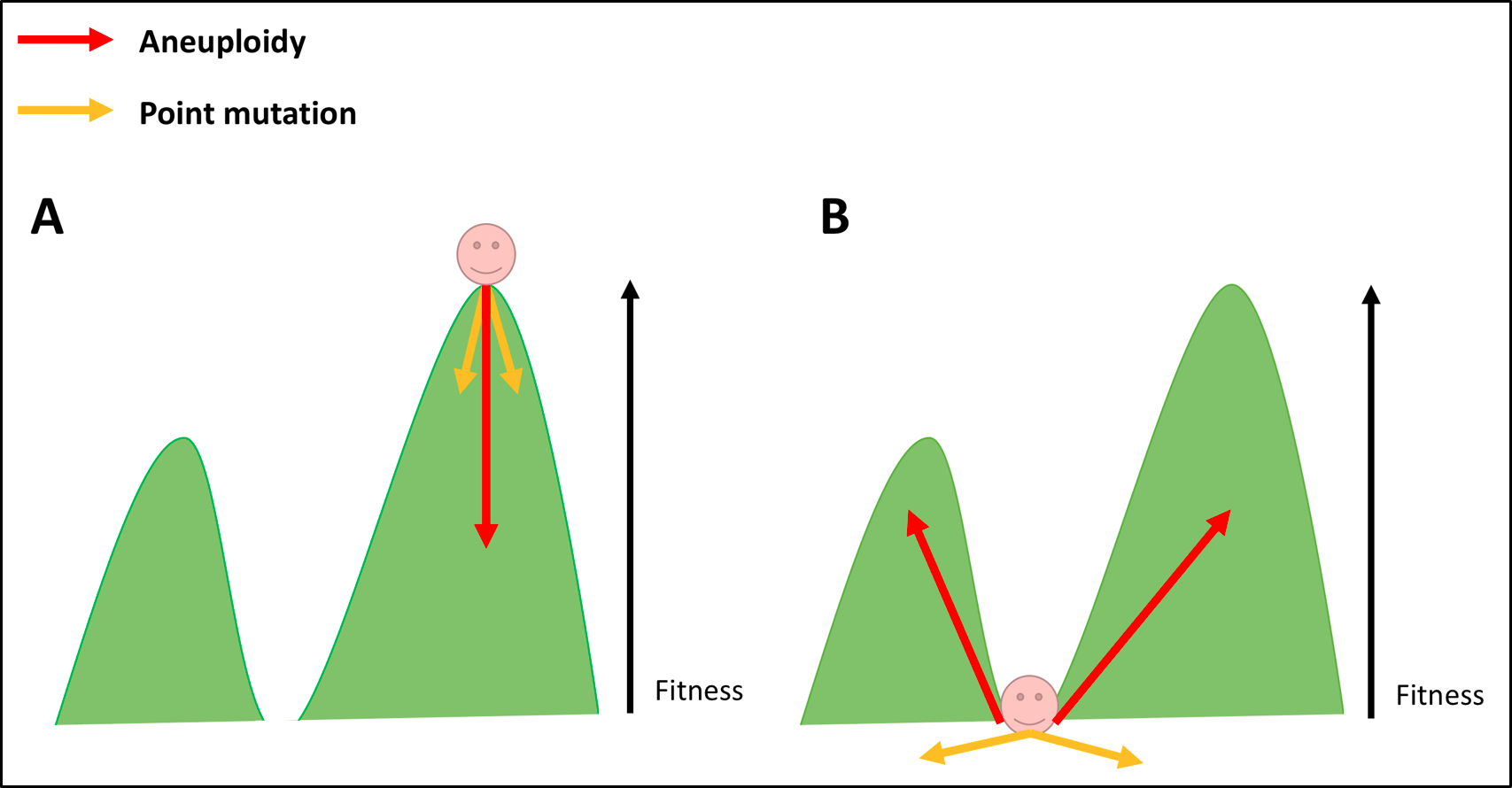
Tumour cells which are well adapted to their surrounding environment are represented in Fig 3A as a hiker atop a ‘fitness peak’. By altering the expression of multiple genes simultaneously, aneuploidy offers these cancer cells a ‘large phenotypic leap’. In Fig 3A, those large leaps are likely to cause cells to ‘fall from the peak’. In such circumstances, aneuploidy is likely to be detrimental to tumorigenesis [30]. Fig 3B represents tumour cells that stand to benefit from aneuploidy. Located in the ‘fitness valley’, cells able to sample the extensive ‘aneuploid landscape’ of genetic variation have a greater chance of acquiring large phenotypic change with immediate selective advantages [26]. It in by this means that aneuploidy is thought to aid metastasizing cells in particular in adapting to a new environment rapidly. Such an event is unlikely to be achieved by single point mutational events represented by the (yellow arrows in Fig 3B) [6][26]. It appears, then, that aneuploidy may be both detrimental or beneficial to cancer cells, depending on the shape of the fitness landscape, the genetic context of the tumour cells, and how well adapted cells are to their surrounding environment [26]. Further research, particularly on metastatic tumours (and the primary counterparts from which they spread), would provide evidence for this theoretical solution to the conundrum.
8. Oversimplification of the Conundrum?
The above model is based on evidence from the discussed model organism studies. Perhaps that is too simplistic a model of aneuploidy in cancer. After all, it would be very rare to find a tumour cell whose karyotype was altered solely by the addition of a single chromosome [39]. Additionally, the model offers no explanation for the difference in frequency of aneuploidy observed in solid tumours in comparison to that of hematological cancers. Perhaps in the case of the latter, a high incidence of VDJ rearrangements negates the need for aneuploidy. Further research in this field might offer new insights into this conundrum.
9. Resolving the Conundrum: Tolerating Aneuploidy?
Even if aneuploidy confers an immediate, large phenotypic leap to aid some cancers, this evolutionary theory model fails to directly address the negative effects of aneuploidy on cellular fitness, (namely reduced cellular proliferation and proteotoxicity), central to this conundrum. Amon and Sheltzer offer a solution; they argue that cells can evolve mechanisms via mutations to tolerate these detrimental effects, the acquisition of which may be enhanced by (aneuploidy associated) genomic instability [4][9]. The logic behind tolerance mechanisms is that alterations in copy number of a single chromosome should have a far greater impact on haploid cells, than on diploid cells \textbf{ [7]}. Evidence as such comes from studies performed on tetraploid yeast cells; a near 1000 fold increase in the rate of chromosome loss or gain can be tolerated without significant impairment on cell cycle proliferation rate, a vast difference from the previously described sensitive effects of aneuploidy on haploid yeast strains [20][40]. These buffering effects of polyploidy might be one mechanism by which cells tolerate the detrimental effects of aneuploidy. But how do cancer cells tolerate aneuploidy associated proteotoxicity? Human aneuploid cells were shown to up-regulate P62 dependent autophagy and lysosomal degradation [22]. Similar findings in MEFs of increased rates of autophagy and elevated basal levels of the Hsp72 chaperone were shown. These mechanisms, if heightened in cancer, may reduce levels of proteotoxicity sufficiently to tolerate the aneuploid state [6]. The concept of tolerance mechanisms may be applied to bridge the gap between aneuploid phenotypes at the cellular level and those at the whole organism level. Constitutionally aneuploid embryos whose cells fail to acquire tolerance mechanisms might suffer the detrimental effects of aneuploidy on cellular fitness to the extent that they are incompatible with life. Spontaneous abortion, as observed, would result [8]. Yet embryos whose cells do acquire tolerance to aneuploidy early in foetal development survive, albeit with the associated detrimental effects on human development and predisposition to cancer. The difficulty of tolerance acquisition, and extent to which the detrimental effects of aneuploidy exist and may be dampened, may add further explanation as to why only aneuploidy of the smallest, gene poor autosomes, or dosage compensated sex chromosomes can be tolerated in humans [30]. Cancer cells, with their highly abnormal genetic background, may have a higher chance of acquiring tolerating mechanisms, or ‘additional oncogenic events’ [4], accounting for the relatively very common and complex karyotypes seen in cancer cells [24].
10. Conclusion and Future Perspectives
Whilst it may seem initially perplexing to place aneuploidy, a condition known to reduce cellular proliferation in untransformed cells, in a disease characterised by its over proliferative capacity, this article offers suggestions of how it might not be such a conundrum after all. Central to this theory is the idea that the consequences of aneuploidy are highly dependent on the genetic and environmental context of a cell [26]. As cancer seems to have exploited aneuploidy for its benefit, so may Medicine do the same [35]. Aneuploidy as a therapeutic target offers broad-spectrum applications for cancer therapy. Currently in clinical trials, chemotherapeutic agents 17-AAG (Hsp90 inhibitor) and AICAR (AMPK activator) exacerbate proteotoxicity and metabolic stress, respectively, to synthetically and selectively induce lethality in aneuploid cancer cells [41][42]. Aneuploidy may be exploited by radiotherapy, too. Bakhoum and colleagues showed that treatment with ionizing radiation leads to ‘mitotic chromosome segregation errors in vivo and long-lasting aneuploidy in tumour derived cell lines’. When this whole-chromosome mis-segregation was experimentally suppressed, orthotopically transplanted human glioblastoma tumours were rendered significantly more resistant to ionizing radiation. This mitotic pathway can be exploited clinically to modulate response to treatment [43]. There is therapeutic promise in aneuploidy for Down syndrome individuals, too, albeit in the distant pipeline. Efforts to exploit Xist sex chromosome dosage compensation machinery as a means of silencing the additional copy of chromosome 21 has shown promise in stem cell models [7]. It will be interesting to see whether this idea can stand the test of time to make its way into the clinic. Aneuploidy is also proving useful in prognostication. Aneuploidy associated miss-segregation has been found to predict enhanced pathological response of rectal adenocarcinoma to chemoradiation therapy. Zaki and colleagues hypothesize that structural chromosomal damage resulting from errors of segregation potentiates the therapeutic targeting of DNA repair machinery [44]. Whilst progress has been made in solving this conundrum, many questions remain to be addressed. Elucidating further mechanistic detail of exactly how aneuploidy may benefit tumorigenesis is key. Determining how the effects of aneuploidy are modulated by cellular genetic and environment factors in cancer and in constitutional aneuploidy will also prove vital [26]. Finally, further evidence for tolerance mechanisms is required to add scientific weight to this theory. The continued development of this field not only offers a fresh perspective on cancer biology and inherited disease, it promises to be an interesting field to follow from a therapeutics perspective, as both a scientist and a clinician.
References
[1] Williams BR, Amon A. Aneuploidy –Cancer’s Fatal Flaw? Cancer Res [Internet]. 2009 Jul 1 [cited 2015 Dec 13];69(13):5289–91.<\p>
[2] Weaver BA, Cleveland DW. Does aneuploidy cause cancer? Curr Opin Cell Biol [Internet]. 2006 Dec [cited 2016 Jan 6];18(6):658–67.<\p>
[3] Segal DJ, McCoy EE. Studies on Down’s syndrome in tissue culture. I. Growth rates and protein contents of fibroblast cultures. J Cell Physiol. 1974 Feb;83(1):85–90.<\p>
[4] Ganmore I, Smooha G, Izraeli S. Constitutional aneuploidy and cancer predisposition. Hum Mol Genet [Internet]. 2009 Apr 15 [cited 2016 Jan 5];18(R1):R84–93.<\p>
[5] Hardy PA, Zacharias H. Reappraisal of the Hansemann-Boveri hypothesis on the origin of tumors. Cell Biol Int. 2005 Dec;29(12):983–92.<\p>
[6] Siegel JJ, Amon A. New Insights into the Troubles of Aneuploidy. Annu Rev Cell Dev Biol [Internet]. 2012 [cited 2015 Dec 13];28:189–214<\p>
[7] Santaguida S, Amon A. Short- and long-term effects of chromosome mis-segregation and aneuploidy. Nat Rev Mol Cell Biol [Internet]. 2015 Aug [cited 2015 Dec 13];16(8):473–85.<\p>
[8] Hassold T, Abruzzo M, Adkins K, Griffin D, Merrill M, Millie E, et al. Human aneuploidy: incidence, origin, and etiology. Environ Mol Mutagen. 1996;28(3):167–75.<\p>
[9] Sheltzer JM, Amon A. The aneuploidy paradox: costs and benefits of an incorrect karyotype. Trends Genet [Internet]. 2011 Nov [cited 2015 Dec 13];27(11):446–53.<\p>
[10] Rasmussen SA, Wong L-YC, Yang Q, May KM, Friedman JM. Population-based analyses of mortality in trisomy 13 and trisomy 18. Pediatrics. 2003 Apr;111(4 Pt 1):777–84.<\p>
[11] Lott IT. Neurological phenotypes for Down syndrome across the life span. Prog Brain Res. 2012;197:101–21.<\p>
[12] Kingsbury MA, Yung YC, Peterson SE, Westra JW, Chun J. Aneuploidy in the normal and diseased brain. Cell Mol Life Sci CMLS. 2006 Nov;63(22):2626–41.<\p>
[13] Cataldo AM, Petanceska S, Peterhoff CM, Terio NB, Epstein CJ, Villar A, et al. App gene dosage modulates endosomal abnormalities of Alzheimer’s disease in a segmental trisomy 16 mouse model of down syndrome. J Neurosci Off J Soc Neurosci. 2003 Jul 30;23(17):6788–92.<\p>
[14] Oromendia AB, Amon A. Aneuploidy: implications for protein homeostasis and disease. Dis Model Mech [Internet]. 2014 Jan 1 [cited 2015 Dec 13];7(1):15–20<\p>
[15] Hasle H. Pattern of malignant disorders in individuals with Down’s syndrome. Lancet Oncol. 2001 Jul;2(7):429–36.<\p>
[16] Sussan TE, Yang A, Li F, Ostrowski MC, Reeves RH. Trisomy represses Apc(Min)-mediated tumours in mouse models of Down’s syndrome. Nature. 2008 Jan 3;451(7174):73–5.<\p>
[17] Baek K-H, Zaslavsky A, Lynch RC, Britt C, Okada Y, Siarey RJ, et al. Down’s syndrome suppression of tumour growth and the role of the calcineurin inhibitor DSCR1. Nature. 2009 Jun 25;459(7250):1126–30.<\p>
[18] Schoemaker MJ, Swerdlow AJ, Higgins CD, Wright AF, Jacobs PA, UK Clinical Cytogenetics Group. Cancer incidence in women with Turner syndrome in Great Britain: a national cohort study. Lancet Oncol. 2008 Mar;9(3):239–46.<\p>
[19] Swerdlow AJ, Schoemaker MJ, Higgins CD, Wright AF, Jacobs PA, UK Clinical Cytogenetics Group. Cancer incidence and mortality in men with Klinefelter syndrome: a cohort study. J Natl Cancer Inst. 2005 Aug 17;97(16):1204–10.<\p>
[20] Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, Dunham MJ, et al. Effects of Aneuploidy on Cellular Physiology and Cell Division in Haploid Yeast. Science [Internet]. 2007 Aug 17 [cited 2015 Dec 15];317(5840):916–24<\p>
[21] Williams BR, Prabhu VR, Hunter KE, Glazier CM, Whittaker CA, Housman DE, et al. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science [Internet]. 2008 Oct 31 [cited 2016 Jan 6];322(5902):703–9<\p>
[22] Stingele S, Stoehr G, Peplowska K, Cox J, Mann M, Storchova Z. Global analysis of genome, transcriptome and proteome reveals the response to aneuploidy in human cells. Mol Syst Biol [Internet]. 2012 Sep 11 [cited 2016 Jan 6];8:608<\p>
[23] Prestel M, Feller C, Becker PB. Dosage compensation and the global re-balancing of aneuploid genomes. Genome Biol [Internet]. 2010 Aug 26 [cited 2015 Dec 18];11(8):216.<\p>
[24] Gordon DJ, Resio B, Pellman D. Causes and consequences of aneuploidy in cancer. Nat Rev Genet [Internet]. 2012 Mar [cited 2015 Dec 15];13(3):189–203.<\p>
[25] Oromendia AB, Dodgson SE, Amon A. Aneuploidy causes proteotoxic stress in yeast. Genes Dev [Internet]. 2012 Dec 15 [cited 2015 Dec 15];26(24):2696–708.<\p>
[26] Pavelka N, Rancati G, Li R. Dr Jekyll and Mr Hyde: role of aneuploidy in cellular adaptation and cancer. Curr Opin Cell Biol [Internet]. 2010 Dec [cited 2015 Dec 13];22(6):809–15.<\p>
[27] Bean J, Brennan C, Shih J-Y, Riely G, Viale A, Wang L, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A. 2007 Dec 26;104(52):20932–7.<\p>
[28] Li M, Fang X, Baker DJ, Guo L, Gao X, Wei Z, et al. The ATM-p53 pathway suppresses aneuploidy-induced tumorigenesis. Proc Natl Acad Sci U S A. 2010 Aug 10;107(32):14188–93.<\p>
[29] Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009 May 22;324(5930):1029–33.<\p>
[30] Sheltzer JM, Blank HM, Pfau SJ, Tange Y, George BM, Humpton TJ, et al. Aneuploidy drives genomic instability in yeast. Science. 2011 Aug 19;333(6045):1026–30.<\p>
[31] Tang Y-C, Amon A. Gene Copy-Number Alterations: A Cost-Benefit Analysis. Cell [Internet]. 2013 Jan 31 [cited 2015 Dec 13];152(3):394–405.<\p>
[32] Sotillo R, Schvartzman J-M, Socci ND, Benezra R. Mad2-induced chromosome instability leads to lung tumour relapse after oncogene withdrawal. Nature. 2010 Mar 18;464(7287):436–40.<\p>
[33] Albertson DG, Collins C, McCormick F, Gray JW. Chromosome aberrations in solid tumors. Nat Genet. 2003 Aug;34(4):369–76.<\p>
[34] Manning AL, Longworth MS, Dyson NJ. Loss of pRB causes centromere dysfunction and chromosomal instability. Genes Dev. 2010 Jul 1;24(13):1364–76.<\p>
[35] Bakhoum SF, Compton DA. Chromosomal instability and cancer: a complex relationship with therapeutic potential. J Clin Invest [Internet]. 2012 Apr 2 [cited 2015 Dec 13];122(4):1138–43.<\p>
[36] Janssen A, Kops GJPL, Medema RH. Elevating the frequency of chromosome mis-segregation as a strategy to kill tumor cells. Proc Natl Acad Sci U S A. 2009 Nov 10;106(45):19108–13.<\p>
[37] Komarova NL, Wodarz D. The optimal rate of chromosome loss for the inactivation of tumor suppressor genes in cancer. Proc Natl Acad Sci U S A. 2004 May 4;101(18):7017–21.<\p>
[38] Kauffman S, Levin S. Towards a general theory of adaptive walks on rugged landscapes. J Theor Biol [Internet]. 1987 Sep 7 [cited 2017 Sep 4];128(1):11–45<\p>
[39] Torres EM, Williams BR, Tang Y-C, Amon A. Thoughts on Aneuploidy. Cold Spring Harb Symp Quant Biol [Internet]. 2010 [cited 2015 Dec 13];75:445–51.<\p>
[40] Storchov\’a Z, Breneman A, Cande J, Dunn J, Burbank K, O’Toole E, et al. Genome-wide genetic analysis of polyploidy in yeast. Nature. 2006 Oct 5;443(7111):541–7.<\p>
[41] Usmani SZ, Bona R, Li Z. 17 AAG for HSP90 inhibition in cancer-from bench to bedside. Curr Mol Med. 2009 Jun;9(5):654–64.<\p>
[42] Tang Y-C, Williams BR, Siegel JJ, Amon A. Identification of aneuploidy-selective antiproliferation compounds. Cell. 2011 Feb 18;144(4):499–512.<\p>
[43] Bakhoum SF, Kabeche L, Wood MD, Laucius CD, Qu D, Laughney AM, et al. Numerical chromosomal instability mediates susceptibility to radiation treatment. Nat Commun. 2015 Jan 21;6:5990.<\p>
[44] Zaki BI, Suriawinata AA, Eastman AR, Garner KM, Bakhoum SF. Chromosomal instability portends superior response of rectal adenocarcinoma to chemoradiation therapy. Cancer. 2014 Jun 1;120(11):1733–42.<\p>
Article photo credit: NIH Image Gallery
- Log in to post comments










