It’s all in the Head?
Abstract
69 year old male with sudden onset headache, deteriorating vision accompanied by features of hypopituitarism.
Clinical Narrative
Presentation
Mr F, a Maltese 69 year old retired electrician living at home with his wife, presented to the Emergency Department with a referral from his ophthalmologist. Mr F presented with a two month history of headaches that had worsened the last two weeks accompanied by a deterioration in peripheral vision over the last week. The headache was described as continuous and generalised, had suddenly increased in severity from 2/10 to 7/10 over the last 2 weeks, and was not affected by posture, movement or time of day. Mr F denied any associated vomiting, drowsiness, neck stiffness, photophobia or rhinorrhoea. Other clinical features noted were fatigue, cold intolerance, decreased need for shaving and decreased libido for the past month. No other constitutional symptoms including weight loss, change in appetite, fevers, or night sweats were reported. No changes in bladder or bowel function.
His past medical history includes Type 2 Diabetes Mellitus, hypertension, and atrial fibrillation, and hence current medication included Metformin, Digoxin, Prazosin and Warfarin. Social history revealed no pets, allergies or recent travel; he occasionally drinks alcohol and has never smoked. Family history was unremarkable.
On examination Mr F was wearing a thick jacket in a warm examination room and appeared comfortable and orientated to time, place and person with a Glasgow Coma Scale of 15/15. He was apyrexial with a blood pressure of 158/86, a regular pulse rate of 84 beats per minute and a respiratory rate of 16 breaths per min.
Full Visual Examination
Pupils equal and reacting to light, there was no nystagmus or diplopia. The remaining cranial nerve examination was normal. Upper and lower limb neurological examination showed normal tone, power, coordination, reflexes and sensation.
Differential Diagnosis
With this presentation of bitemporal hemaniopia, headache and associated features of hypopituitarism, a pituitary adenoma was the main differential.
Other pituitary masses to think about include crangiopharyngiomas: these are likely to be suprasellar and are the most common sella region tumour in children [1]. Rarer masses include meningiomas, optic nerve and hypothalamic gliomas, chordomas, germinomas and pinealomas [2]. Further down the list, cystic lesions such as Rathke cleft cysts and arachnoid cysts are a possibility. There would also be a need to exclude carotid artery aneurysm presenting with these symptoms, however it seems unlikely as there were no cavernous sinus cranial nerve abnormalities.
The time course of the sudden deterioration was also taken into consideration; could this be something more serious like pituitary apoplexy?
With these in mind the investigations requested included bloods to look at endocrine function, MRI to determine whether a mass was present and formal visual field testing (automated computed perimetry shown above).
Results Of Investigations
Full blood count, urea and electrolytes, liver function tests, and blood glucose were normal. Clotting studies revealed a raised INR of 4.0. Thyroid function tests were abnormal with low free T3 (2.7 [3.5-6.5]pmol/L) and T4 (8.8 [9.0-26.0] pMol/L) and normal TSH (0.6 [0.1-4.0] mIU/L). Other hormonal imbalances included low LH (0.5 [1.5-9.3]IU/L), low testosterone (0.9 [7-28]IU/L). Noon plasma cortisol was on the lower end of normal (122 [120-650]nmol/L), and prolactin was normal (326 [45-375]mIU/L). This generated an overall picture of secondary hypothyroidism and secondary hypogonadism.
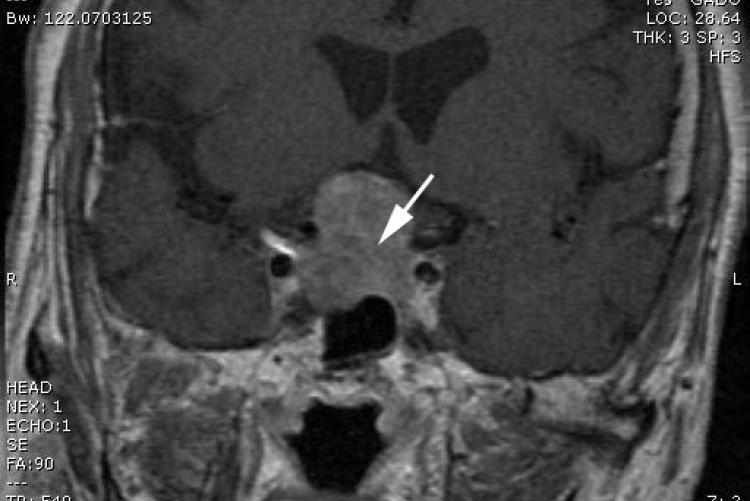
The MRI (figure 1) showed a mass centred within the sella turcica measuring 3.3 x 2.5 x 2cm. The mass expands the sella and projects into the suprasellar cistern. It compressed the optic chiasm, but there was no evidence of cavernous sinus involvement with normal cavernous carotid flow voids. The mass demonstrated mild patchy contrast enhancement. No other abnormalities and no ventricular involvement seen.
Treatment
With these findings and the sudden increase in severity of Mr F’s headache and visual deterioration leading him to present acutely, on a background of a longer hypopituitary picture suggested the clinical diagnosis of pituitary tumour apoplexy. Urgent steroids (hydrocortisone) were started and endoscopic transphenoidal surgery was performed to remove the pituitary macroadenoma the next day. Figure 2 shows the biopsy, demonstrating a null cell adenoma with focal haemorrhagic infarction.
For completion I have included Mr F’s greatly improved 4 weeks post operative visual fields (table 2). Postoperatively he is well, having regular follow up and is taking thyroxine replacement.
Discussion
Pituitary apoplexy is rare but is important to think about because it is potentially sight- and life-threatening.
It is characterised by a sudden onset headache, visual deterioration, vomiting and decreased consciousness as well as ocular palsies caused by the rapid enlargement from a bleed into the tumour [3]. It also causes impaired pituitary function.
It is unclear what precipitates it but some predisposing factors include hypertension, anticoagulation, closed head trauma, cardiac surgery, treatment with dopamine agonists, previous pituitary irradiation or stimulation testing and pregnancy [4].
Current UK guidelines require multidisciplinary team involvement once the diagnosis has been made, comprising, amongst others, a neurosurgeon and an endocrinologist. The main reason for mortality associated with pituitary apoplexy is the result of acute secondary adrenal insufficiency. Thus prompt high dose corticosteroid replacement, even when waiting for confirmatory investigations is necessary [5].
It is however unclear in the literature at what stage surgical decompression should be employed. Some authors suggest that early surgery will have better outcomes with regards to vision and endocrine function post operatively, whereas others have suggested that there is no difference between patients managed conservatively with high dose hydrocortisone and those with early surgical resection [6,7]. It is difficult to evaluate this and few randomised control trials have been done due to the rarity of the condition. The observational studies which have been done lack appropriately matched patients; those with worse visual symptoms are often treated surgically whilst those that show little neurological deterioration are managed conservatively, and the outcomes compared It is clear there needs to be further evidence to suggest what the best approach to management should be.
Take Home Messages
1) Reasons for clinical symptoms arising from a pituitary tumour can be divided into three: the local anatomical effects of the tumour; the effects of decreased hormone production by the remaining pituitary tissue; and symptoms related to excess hormone secretion by a hormonally active tumour, depending on the particular hormone secreted.
2) Have a high index of suspicion for pituitary apoplexy with patients with acute severe headache and visual deterioration, especially ocular palsies.
3) Start high dose hydrocortisone if suspicious – can be potentially sight and life saving.
4) Patients will need to be followed up post surgery, with some patients requiring long term hormonal therapy. Communication and education about this is necessary – we required a Maltese interpreter to discuss this with Mr F and Mrs F to ensure they understood the importance of this.
Acknowledgements
Many thanks to Mr James King, Consultant Neurosurgeon at The Royal Melbourne Hospital, for his help discussing the case and obtaining the images.
1. Kumar and Clark. Clinical Medicine. Seventh Edition.
2. Professor Andrew Kaye. Essential Neurosurgery. Third Edition.
3. Vanderpump M, Higgens C, Wass JAH. UK guidelines for the management of pituitary apoplexy a rare but potentially fatal medical emergency. Emerg Med J. 2011 Jan 7;28(7):550–1. DOI: 10.1136/emj.2010.106898
4. Murad-Kejbou S, Eggenberger E. Pituitary apoplexy: evaluation, management, and prognosis. Curr Opin Ophthalmol. 2009 Nov;20(6):456–61. DOI: 10.1097/ICU.0b013e3283319061
5. Pituitary Apoplexy. 2013 Jan 25 [cited 2013 Aug 26]; Available from:http://emedicine.medscape.com/article/1198279-overview
6. Rajasekaran S, Vanderpump M, Baldeweg S, Drake W, Reddy N, Lanyon M, et al. UK guidelines for the management of pituitary apoplexy. Clin Endocrinol (Oxf). 2011;74(1):9–20. DOI: 10.1111/j.1365-2265.2010.03913.x
7. Sibal L, Ball SG, Connolly V, James RA, Kane P, Kelly WF, et al. Pituitary apoplexy: a review of clinical presentation, management and outcome in 45 cases. Pituitary. 2004;7(3):157–63. DOI: 10.1007/s11102-005-1050-3
- Log in to post comments










